
Apert Syndrome: Practice Essentials, Pathophysiology, Epidemiology
Apert syndrome is a rare autosomal dominant disorder characterized by craniosynostosis, craniofacial anomalies, and severe symmetrical syndactyly (cutaneous and bony fusion) of the hands and feet (see the images below). It is probably the most familiar and best-described type of acrocephalosyndactyly. Reproductive fitness is low, and more than 98% of cases arise by new mutation.The syndrome is named for the French physician who described the syndrome acrocephalosyndactylia in 1906.
[1]
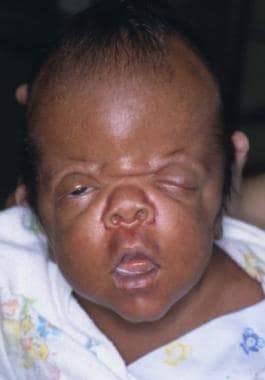
An infant with Apert syndrome is shown. Note the characteristic ocular hypertelorism, down-slanting palpebral fissures, proptotic eyes, horizontal groove above the supraorbital ridge, break of the eyebrows’ continuity, depressed nasal bridge, and short, wide nose with bulbous tip.
View Media Gallery
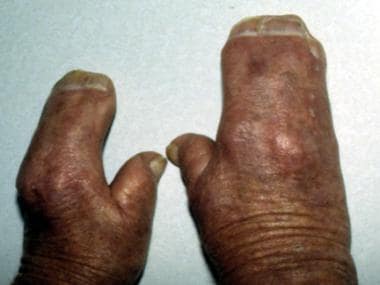
Note the mitten appearance of the hands with syndactyly involving the second, third, fourth, and fifth fingers. This patient also has characteristic concave palms, hitchhiker posture (radial deviation) of the short broad thumbs, and contiguous nail beds (synonychia).
View Media Gallery
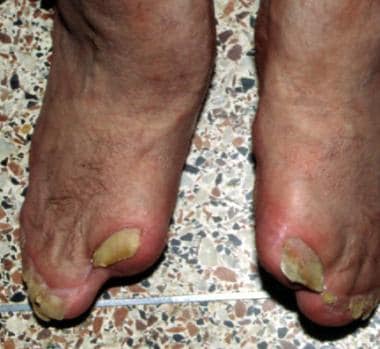
Note the socklike appearance of the feet with syndactyly involving the second, third, fourth, and fifth toes. The patient also has contiguous nail beds (synonychia).
View Media Gallery
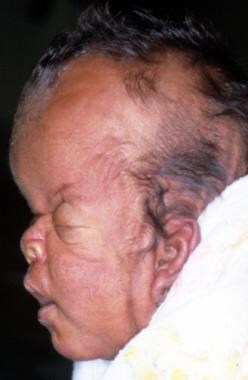
In this profile photo, turribrachycephaly (high prominent forehead), proptosis, a depressed nasal bridge, a short nose, and low-set ears are prominent.
View Media Gallery
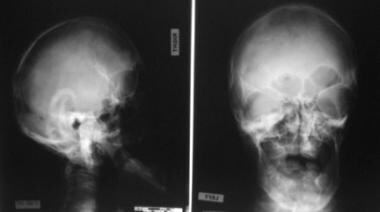
This radiograph demonstrates turribrachycephaly, shallow orbits, ocular hypertelorism, and a hypoplastic maxilla.
View Media Gallery
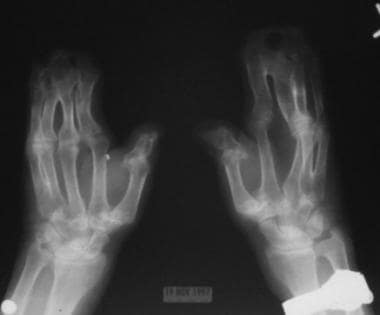
Note the osseous syndactyly involving the second, third, fourth, and fifth fingers; multiple synostosis involving the distal phalanges and proximal fourth and fifth metacarpals; symphalangism of the interphalangeal joints; shortening and radial deviation of the distal phalanx; and the delta-shaped deformity of proximal phalanx of the thumbs.
View Media Gallery
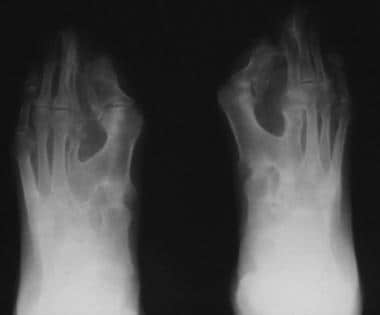
Note the osseous syndactyly, fusion of the interphalangeal joints, synostosis involving the proximal first and second metatarsals, and the partially duplicated and delta-shaped proximal phalanx of the great toes.
View Media Gallery
Signs and symptoms of Apert syndrome
With craniosynostosis, coronal sutures most commonly are involved, resulting in acrocephaly, brachycephaly, turribrachycephaly, flat occiput, and high prominent forehead.
Patients have apparent low-set ears, with occasional conductive hearing loss and congenital fixation of the stapedial footplate.
The eyes exhibit down-slanting palpebral fissures, hypertelorism, shallow orbits, proptosis, exophthalmos, strabismus, amblyopia, optic atrophy, and, rarely, luxation of the eye globes, keratoconus, ectopic lentis, congenital glaucoma, lack of pigment in the fundi with occasional papilledema, and preventable vision loss or blindness.
The nose has a markedly depressed nasal bridge. It is short and wide, with a bulbous tip, parrot-beaked appearance, and choanal stenosis or atresia.
The mouth area has a prominent mandible, down-turned corners, a high arched palate, a bifid uvula, and a cleft palate.
The upper limbs are more severely affected than lower limbs. Coalition of distal phalanges and synonychia found in the hands are never present in the feet. The glenohumeral joint and proximal humerus are more severely affected than the pelvic girdle and femur. The elbow is much less severely involved than the proximal portion of the upper limb.
Intelligence varies in persons with Apert syndrome from normal to mental deficiency, although a significant number of patients have mental retardation.
Apert syndrome is also characterized by cutaneous, cardiovascular, genitourinary, gastrointestinal, and respiratory disorders.
Workup in Apert syndrome
Regarding the molecular analysis of Apert syndrome, it is known that more than 98% of cases are caused by specific missense substitution mutations involving adjacent amino acids (Ser252Trp, Ser252Phe, or Pro253Arg) in exon 7 of FGFR2.
Imaging studies in Apert syndrome include the following:
-
Skull radiography
-
Spinal radiography
-
Limb radiography
-
Hand radiography
-
Foot radiography
-
Computed tomography (CT) scanning – CT scanning with comparative three-dimensional reconstruction analysis of the calvaria and cranial bases has become the most useful radiologic examination in identifying skull shape and the presence or absence of involved sutures
-
[2, 3]
Magnetic resonance imaging (MRI) – MRI reveals the anatomy of the soft-tissue structures and associated brain abnormalities (ie, nonprogressive ventriculomegaly; hydrocephalus; complete or partial absence of the septum pellucidum; absence of septal leaflets; and thinning, deficiency, or agenesis of the corpus callosum)
Management of Apert syndrome
Surgical management of Apert syndrome can include the following:
-
In severe cases, lateral or medial tarsorrhaphy to protect the corneas
-
In rare cases, orotracheal intubation to manage upper airway obstruction during the neonatal period
-
Tracheostomy in children severely affected by sleep apnea
-
Bilateral myringotomy and placement of ventilation tubes to manage chronic middle ear effusion associated with bilateral conductive hearing deficit
-
Cranial surgery to remove synostotic sutures; reshape the calvaria; allow more normal cranial development to proceed with respect to shape, volume, and bone quality; and relieve increased intracranial pressure
-
Orbital surgery to correct ocular proptosis, reduce increased interorbital distance (hypertelorism), and correct increased interior malrotation
-
Nasal surgery to correct the excessively obtuse nasofrontal angle, flat nasal dorsum, and ptotic nasal tip, in infants and children, and to reduce the nasal tip bulk in teenagers and adults
-
Midfacial surgery to normalize midface appearance, expand the inferior orbit, volumetrically expand the nasal and nasopharyngeal airways, and establish a normal dentoskeletal relationship
-
Mandibular osteotomy to improve dentoskeletal relations for masticatory and aesthetic benefit




